
Radiologic diagnosis in multiple endocrine neoplasia type 1 syndrome. A case study based on the medical history of a father and his two children.
DOI:
https://doi.org/10.3941/jrcr.4668Keywords:
multiple endocrine neoplasia syndrome type 1, neuroendocrine tumors, adenoma, MEN1 syndrome, Wermer syndromeAbstract
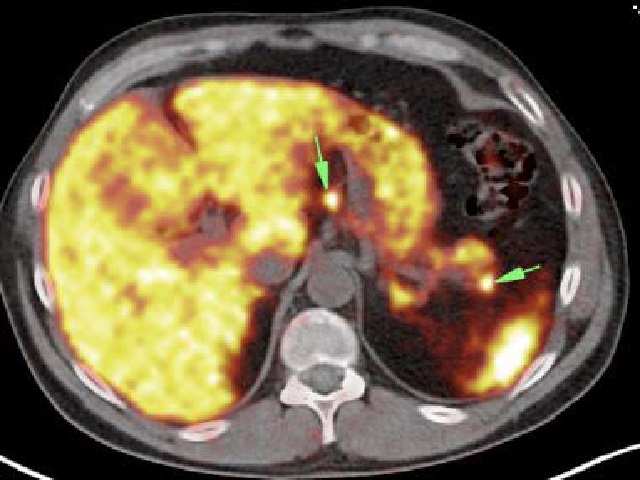
Multiple endocrine neoplasia syndrome type 1 (MEN1) is an autosomal dominant inherited disease. Typical tumours present in patients are parathyroid adenomas, pancreatic neuroendocrine tumour and pituitary adenoma, which may be accompanied by other tumours. The aim of this study was to analyse the clinical cases of a family – a 49-year-old father, a 24-year-old son and a 20-year-old daughter – for the diagnosis of MEN1 syndrome, after confirming the presence of MEN1 in the father. In each of the patients, a mutation in one allele of the MEN1 gene was detected in a heterozygous pattern, which confirmed the diagnosis of endocrine adenomatosis type 1 syndrome. Prior to the diagnosis, none of the children presented with distressing clinical symptoms, daaespite the presence of numerous lesions visualised by radiological examinations, which obtained different histopathological diagnoses. Radiological screening of patients after treatment allows early detection of new lesions, which improves prognosis.
Downloads
Published
Issue
Section
License
Copyright (c) 2024 Journal of Radiology Case Reports

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
The publisher holds the copyright to the published articles and contents. However, the articles in this journal are open-access articles distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 License, which permits reproduction and distribution, provided the original work is properly cited. The publisher and author have the right to use the text, images and other multimedia contents from the submitted work for further usage in affiliated programs. Commercial use and derivative works are not permitted, unless explicitly allowed by the publisher.


